If you have just heard the words “sickle cell disease” at the antenatal clinic or in a family conversation, you probably have one question burning: is it all the same thing, or are there different kinds? The answer matters.
There are several types of sickle cell disease, and they do not all behave the same way in the body. Knowing which type runs in your family changes how you plan, how you prepare, and — most of all — how you raise a child to thrive.
What this article covers
- First, the basics: what sickle cell disease actually is
- Type 1 — HbSS (sickle cell anaemia): the most common form in Ghana
- Type 2 — HbSC: common, often milder, but not harmless
- Type 3 — HbS beta-thalassaemia: rarer, two sub-types
- Type 4 — Rare and compound forms
- How the types are inherited (with a diagram)
- What to ask at your next clinic visit
- Frequently asked questions
First, the basics: what sickle cell disease actually is
Sickle cell disease is an inherited condition that affects haemoglobin — the protein inside red blood cells that carries oxygen around the body.
In a healthy person, red blood cells are smooth, round, and flexible. They glide through blood vessels easily, even the very narrow ones.
In sickle cell disease, the haemoglobin is different. Under certain conditions — when oxygen is low, when the body is cold, dehydrated, or stressed — the red blood cells stiffen and curl into a sickle shape (like a half-moon, or the blade of a farming sickle).
These rigid cells get stuck in blood vessels, block blood flow, and cause the deep, sudden pain that doctors call a crisis.
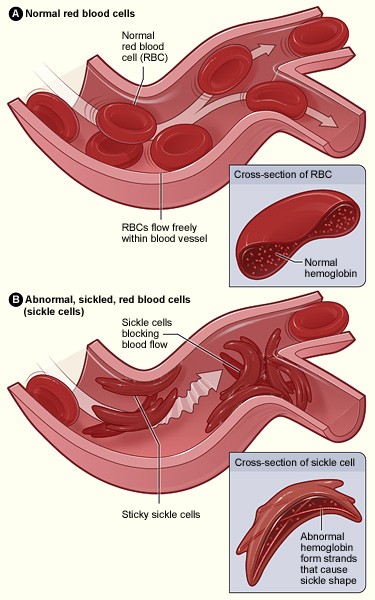
Normal red blood cells (top) flow freely. Sickle cells (bottom) block blood vessels and cause pain. Image: U.S. National Heart, Lung, and Blood Institute (public domain).
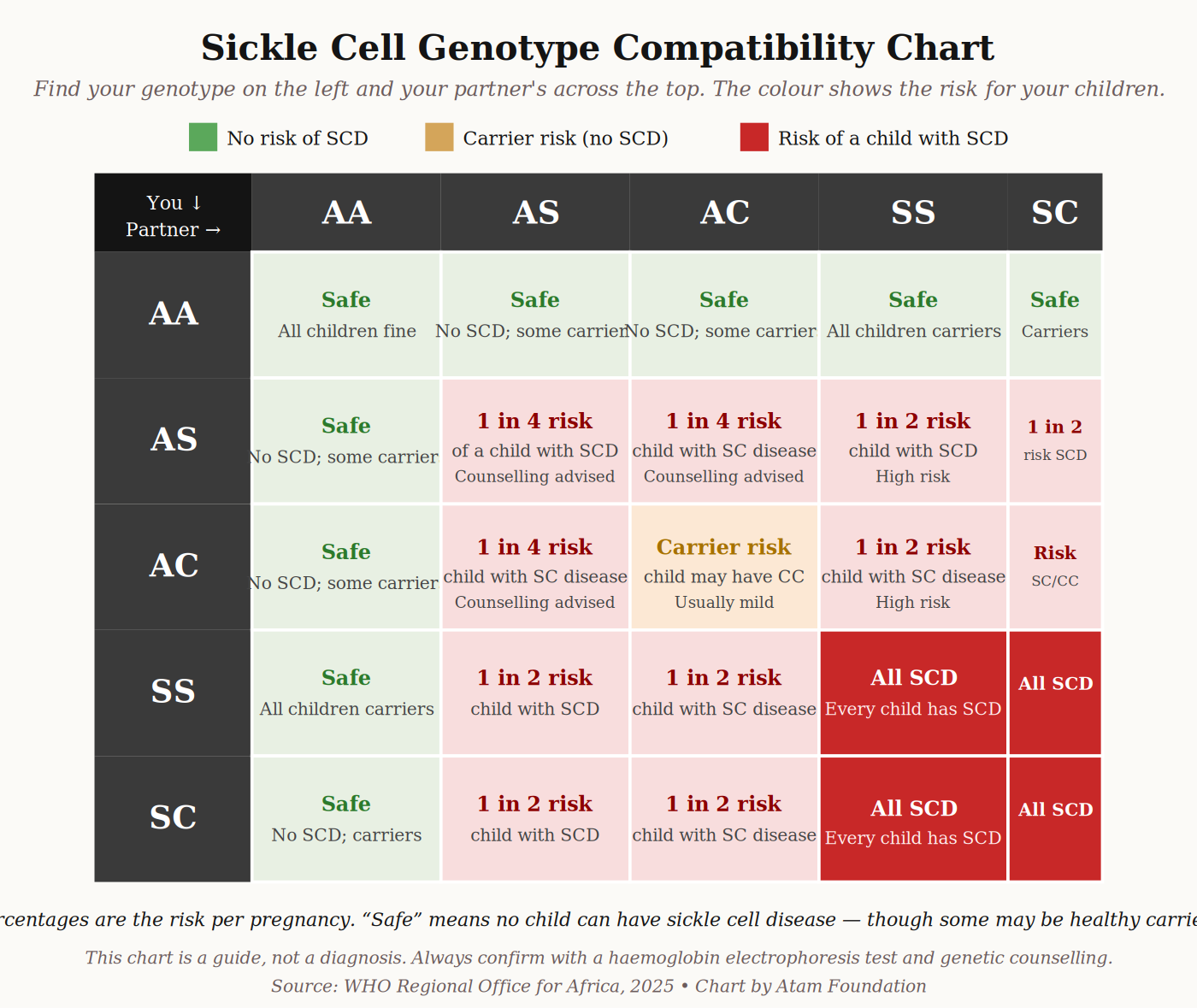
The single letter that decides whether someone has sickle cell disease is S. Each of us inherits two haemoglobin genes — one from our mother, one from our father.
The healthy version is called A. The sickle version is called S. There is also a less common variant called C, and another called β-thalassaemia (sometimes written β-thal).
The type of sickle cell disease you have depends on which two of these you inherited.
| The Ghana picture About 2% of Ghanaian newborns — roughly 15,000 babies every year — are born with sickle cell disease. The Ghana Institute of Clinical Genetics at Korle-Bu has over 27,000 registered patients, with most being HbSS or HbSC. Sources: Asare et al., Advances in Haematology, 2018 — Korle-Bu cohort study. Korle-Bu Teaching Hospital, 50th anniversary report, 2025. |
Type 1 — HbSS (sickle cell anaemia): the most common form
HbSS — also written Hb SS, sickle cell anaemia, or SS disease — happens when a child inherits the S gene from both parents. This is the most common type of sickle cell disease worldwide, and the most common in Ghana. In the Korle-Bu adult cohort, HbSS accounts for around 56% of patients.
What HbSS feels like
HbSS is generally the most severe form. Children with HbSS may begin showing symptoms as early as 4–6 months of age, when their fetal haemoglobin (which protects them in the womb and early infancy) is replaced by the adult version that carries the sickle mutation. Common signs include:
- Deep pain in the hands, feet, back, chest, or abdomen — sometimes lasting days
- Tiredness, paleness, and yellowing of the eyes (due to anaemia)
- Slower growth in early childhood
- Frequent infections — pneumonia, malaria, urinary infections
- Swelling of the hands and feet in babies (dactylitis)
With modern care — folic acid, hydroxyurea, penicillin for young children, prompt malaria treatment, and routine vaccinations — many people with HbSS in Ghana live full adult lives. The World Health Organization notes that hydroxyurea is one of the most effective medicines available for HbSS, and is increasingly available across Africa.
Type 2 — HbSC: common in West Africa, often milder, but not harmless
HbSC — also written Hb SC — happens when a child inherits one S gene and one C gene. The C variant is especially common in West Africa, and almost exclusively found in people of West African ancestry — Ghanaians, Nigerians, and Burkinabè especially. In the Korle-Bu adult cohort, around 40% of patients have HbSC.
What HbSC feels like
HbSC is generally considered milder than HbSS — but that word “milder” needs careful handling. People with HbSC often have less severe anaemia and may grow up without realising they have the condition.
Some are diagnosed only as adults, sometimes after a crisis triggered by pregnancy, surgery, or long-haul flying.
But “milder” does not mean “safe.” People with HbSC face their own pattern of complications:
- Higher risk of eye problems (sickle cell retinopathy) — regular eye check-ups are essential
- Higher risk of avascular necrosis — bone damage, particularly in the hip
- Sudden, severe pain crises that can be just as intense as in HbSS
- Increased complications during pregnancy
| Important — HbSC needs care too If you or your child has been diagnosed with HbSC, do not assume regular check-ups are optional. The complications are different from HbSS, but they are real. Eye examinations every 1–2 years are particularly important from school age onwards. Talk to your doctor about a personalised monitoring plan. |
Type 3 — HbS beta-thalassaemia: rarer, with two sub-types
HbS beta-thalassaemia (HbS β-thal) happens when a child inherits one S gene and one β-thalassaemia gene. β-thalassaemia is a separate haemoglobin disorder more common in the Mediterranean, Middle East, and parts of Asia — but it does occur in Ghana, particularly in families with mixed heritage. There are two sub-types, and the difference matters:
HbS β⁰-thalassaemia (“beta-zero”)
In this form, the β-thalassaemia gene produces no normal haemoglobin at all. The body relies entirely on the sickle version. Clinically, HbS β⁰-thalassaemia behaves almost exactly like HbSS — same severity, same level of care required.
HbS β⁺-thalassaemia (“beta-plus”)
In this form, the β-thalassaemia gene still produces some normal haemoglobin — just less than usual. The body has a partial supply of healthy haemoglobin to fall back on.
As a result, HbS β⁺-thalassaemia is usually milder, sometimes resembling HbSC more than HbSS.
Both sub-types are confirmed through a specialised blood test called haemoglobin electrophoresis — available at Korle-Bu Teaching Hospital, Komfo Anokye Teaching Hospital (KATH) in Kumasi, and a growing number of regional hospitals.
Type 4 — Rare and compound forms
A small number of people inherit S together with another rare haemoglobin variant — for example, HbSD, HbSE, or HbSO-Arab.
These compound forms are uncommon in Ghana but do appear in family histories with diverse heritage. Their severity varies.
If your clinic mentions one of these letter combinations to you, ask for a printed explanation — and bring it to Atam Foundation if you would like a second, plain-language walk-through. We are here for exactly this reason.
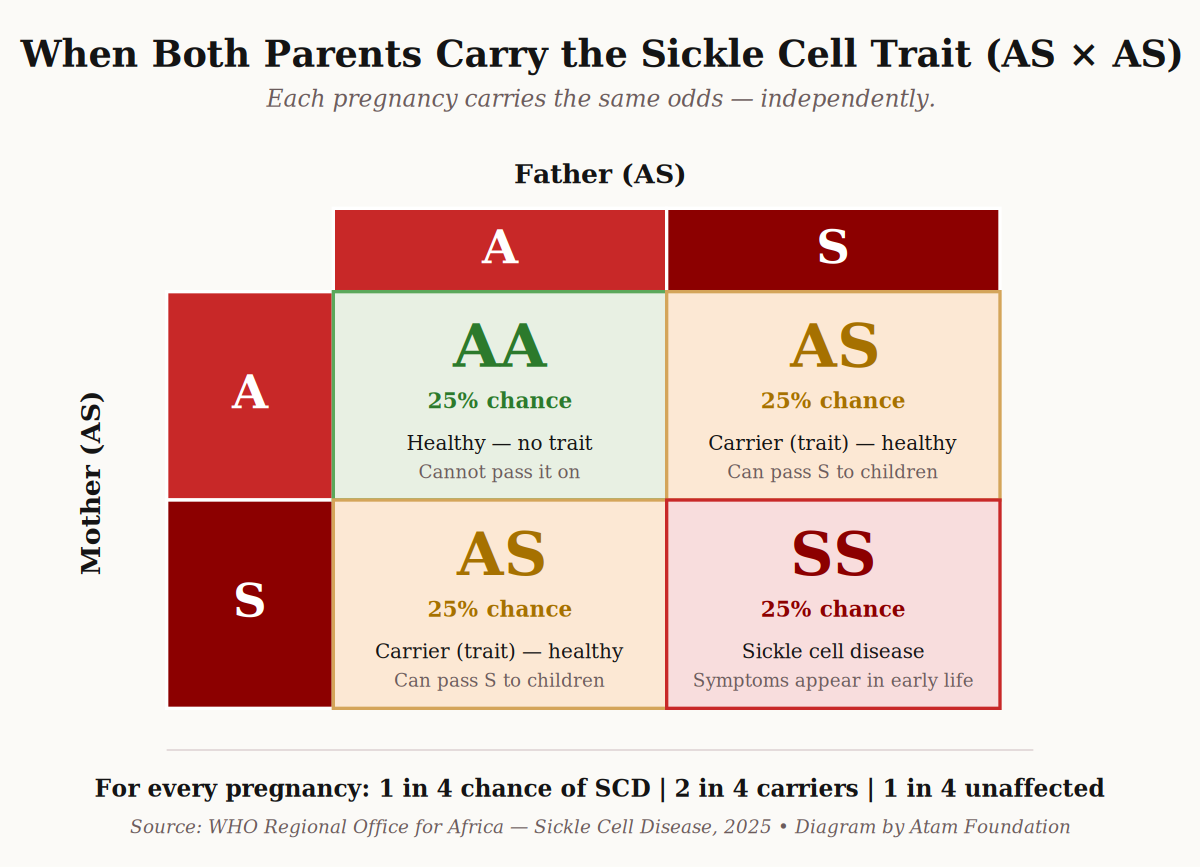
How the types are inherited — the diagram every family should see
Sickle cell disease is not contagious. You cannot catch it. It is inherited — passed from parents to child through the genes they carry.
The key idea: a child can only have sickle cell disease if both parents carry at least one S, C, or β-thal gene. If only one parent carries it, the child can become a carrier (trait), but will not have the disease.
Here is what happens when both parents carry the sickle cell trait (AS):
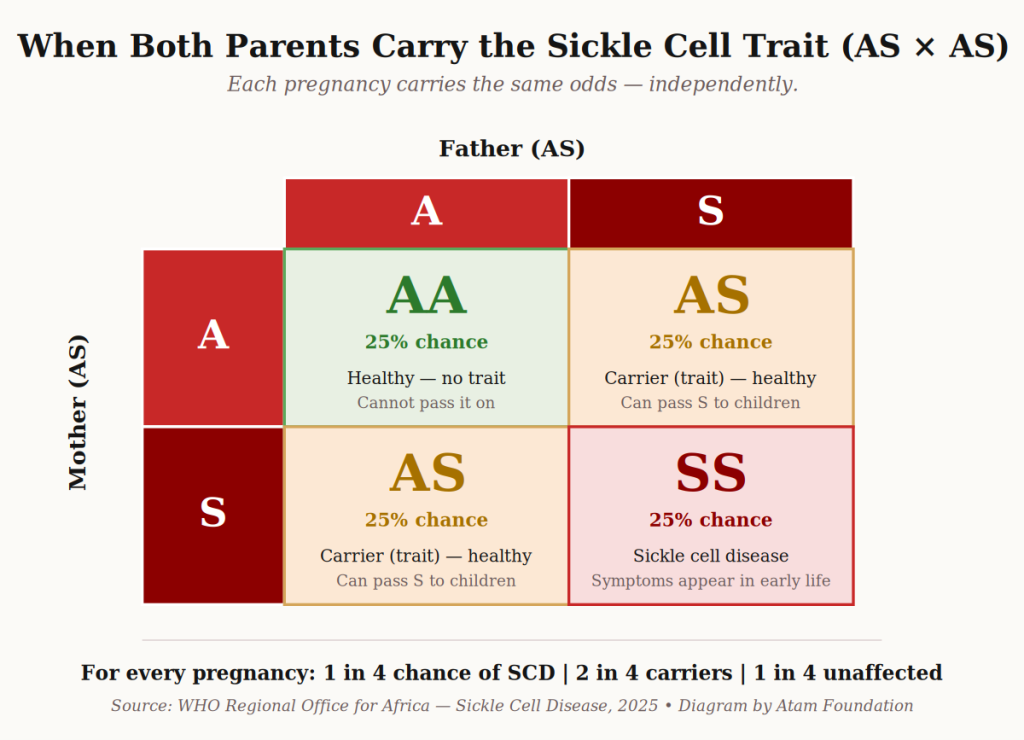
When two parents with sickle cell trait (AS) have a child, every pregnancy carries the same odds. Diagram: Atam Foundation.
This is why premarital screening matters so deeply in Ghana — and why we will dedicate a whole article to it later in this series.
A simple, affordable blood test before marriage tells a couple exactly what genes they each carry, and what the odds are for their future children. It is not about fear; it is about being prepared.

Korle-Bu Teaching Hospital in Accra — home of the Ghana Institute of Clinical Genetics, the country’s premier sickle cell centre. Image: Wikimedia Commons, CC BY-SA 4.0.
What to ask at your next clinic visit
Whether you are getting tested for the first time, expecting a baby, or supporting a child already diagnosed, take this short list with you:
- What exactly is my genotype, or my child’s genotype?
Ask for the printed result. “SS,” “SC,” and “S β-thal” all sound similar but mean different things.
- Which type of sickle cell disease is this?
If it is HbS β-thalassaemia, ask whether it is β⁰ or β⁺ — it makes a real difference.
- What are the most likely complications I should watch for?
Different types have different patterns. Eye checks matter more in HbSC; infection prevention matters more in HbSS young children.
- What is the plan for follow-up?
How often do we come back? Which clinic? Who is the consultant?
- Is hydroxyurea right for us?
It is not for everyone, but it changes lives for many. Ask whether it is being considered.
- Where can we get support beyond this clinic?
This is where organisations like Atam Foundation come in — for community, for information, and for the conversations that take longer than a clinic visit.
Frequently asked questions
Is sickle cell disease curable?
There is one potentially curative treatment — a bone marrow (stem cell) transplant — but it is complex, expensive, and not widely available in Ghana today. For most Ghanaian families, the focus is on excellent management: preventing crises, treating infections promptly, and using medicines like hydroxyurea where appropriate. Gene therapy is emerging globally but is not yet available in West Africa.
Can someone with sickle cell trait (AS) get the disease later in life?
No. The trait is not the disease. A person with AS carries one sickle gene but produces enough normal haemoglobin to live a healthy life. Trait does not “turn into” disease. The only time it matters medically is during family planning.
My child has HbSC and the doctor says it is mild. Should I still worry?
Worry is the wrong word — but stay alert. HbSC is generally milder than HbSS, but it has its own pattern of complications, particularly affecting the eyes and joints. Regular check-ups are essential, even if your child seems perfectly well.
How is sickle cell disease diagnosed in Ghana?
Through a blood test called haemoglobin electrophoresis. It is available at major teaching hospitals — Korle-Bu in Accra, KATH in Kumasi — and at many regional hospitals and private labs. Newborn screening is expanding but not yet universal in Ghana.
Can people with sickle cell disease have children?
Yes. Many people with sickle cell disease in Ghana have healthy pregnancies and children. Pregnancy carries additional risks and needs careful management by a haematologist and obstetrician working together, but it is absolutely possible. We will cover this in detail in a later article.
A final word from Atam Foundation
If this article has answered even one question that was keeping you awake, it has done its job. But you should not have to navigate this alone.
Atam Foundation exists for the families who are still figuring this out. We run educational sessions in Accra and around Ghana, share resources in English and Twi, and connect families with each other and with specialist care.
Whatever your situation — newly diagnosed, planning a family, supporting a relative — we would like to hear from you.